
编辑:医学喵记事
文字:医学喵记事
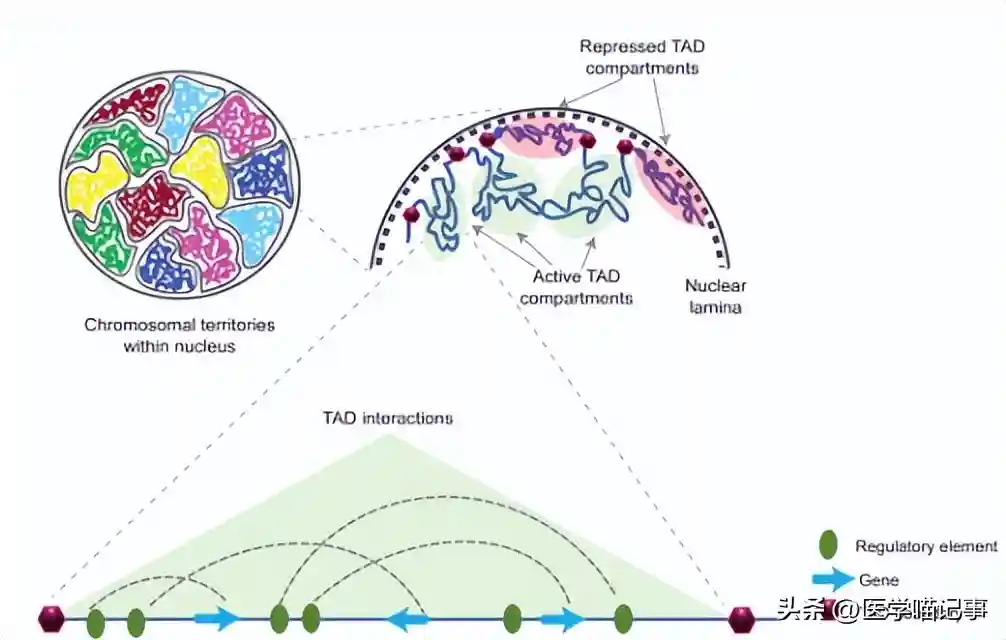
在现代的基因工程研究中,3D基因组拓扑技术的地位在基因调控、基因组组装和基因组重排图谱研究中日益重要。
除了传统的连接法(如Hi-C)外,我们团队开发了一种称为Multiple-GAM的新技术,用于更快、更实惠地进行基因组架构作图(GAM)。
让我们能以更低的成本和时间,实现绘制全基因组范围内的染色质接触图。
在本篇论文中,我们团队主要的实验方式是通过在小鼠胚胎干细胞中比较Multiple-GAM和Hi-C细胞的表达。
一、实验背景介绍一直以来,大部分基因工程团队研究基因调控、基因组组装和基因组构象的时候,采用的全基因组方法都是ChIP-seq、ATAC-seq等方式,3D基因组构象的技术(如Hi-C)已经成为常规模式。
然而,由于细胞的多样性,使得这类标准的研究方式没办法覆盖一些较为罕见的细胞类型。
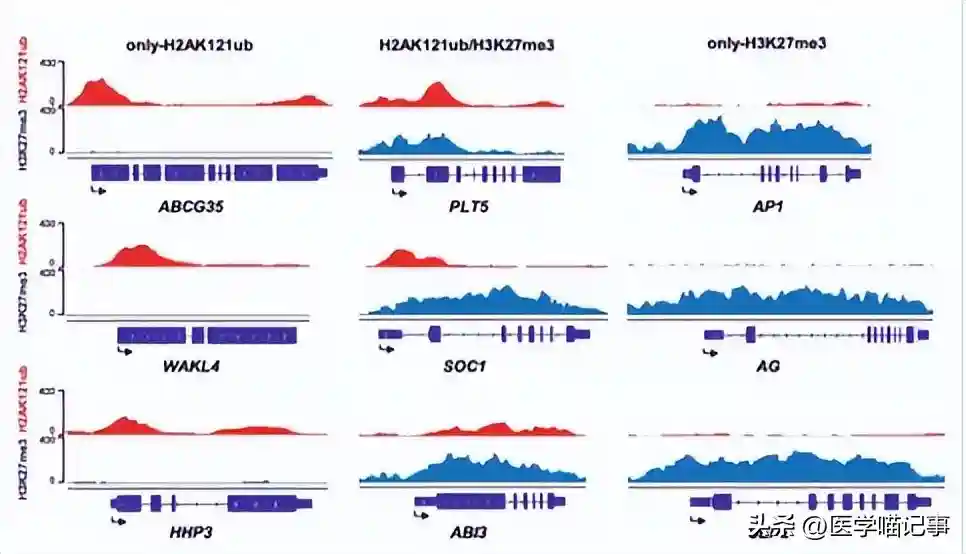
为了解决这种情况,我们团队开发了一种名为Multiple-GAM的技术,用于3D基因组架构作图(GAM),该方法更快、更实惠,无需连接,就能够绘制全基因组范围内的染色质接触图。
为了改进Multiple-GAM,我们进行了一系列重要改进。
首先,通过多重GAM,将多个核图谱添加到单个管中,从而减少了测序的劳动力和成本。
其次,优化了DNA提取方案,实现与液体分配机器人兼容,进一步减少了时间和试剂成本。
第三,扩展了用于分析GAM数据的统计模型,包括适用于多个核图谱的情况。
最后,将GAM数据集从小鼠胚胎干细胞扩展到1,250个细胞,与Hi-C进行了比较。
结果表明,GAM方法与Hi-C相比,能够同样检测到许多接触,并且能够识别特定于方法的接触,特别是涉及三个或更多基因组元件之间同时关联的接触。
GAM是一种通用的染色质接触图谱绘制方法,相对于Hi-C具有多种优势。
我们还提供了一个设计GAM实验的框架,可在最大程度上减少数据收集工作。
二、实验材料备制2.1Multiplex-GAM的多核设计降低实验成本根据我们团队的研究发现,在每个核图谱从不同的细胞核分离到单个PCR管中,特定基因组位点在同一核谱中一起发现的次数是它们在原始细胞群中物理接近度的衡量标准,高共分离值表明这些区域是空间接近。
这说明每个核谱仅包含约5%的基因组,不同染色体上的基因座同时出现在不到1%的核谱中。
因此,我们推断,将多个核谱组合到单个测序文库中不会降低我们区分相互作用和非相互作用位点的能力。
为了测试这个想法,我们使用了单独测序的481个单核谱(481×1NP)数据集,其中包含408个先前发布的样本加上73个额外的单核谱(1NP)数据集。
同时,我们模拟了两个或三个核谱(2NP或3NP)的多重测序,我们组合了480个单核谱数据集,并分别生成了240个或160个包含两个或三个核谱的计算机GAM样本。
然后,我们从这些模拟的Multiple-GAM数据集中重新计算了共分离矩阵,发现这些矩阵在视觉上高度相似并且高度相关。
为了正式了解多重GAM实验设计中包含多个核特征的效果并优化我们的实验参数,我们扩展了SLICE。
这是之前开发的统计工具,用于从GAM数据中的基因座共分离推断非随机DNA相互作用概率.SLICE现在考虑每个GAM样本的核剖面数量、核椭圆率和核剖面厚度的影响。
为了确定在小鼠胚胎干细胞中收集多重GAM数据集的最佳参数,我们应用更新的SLICE模型来估计最小试管数量(m*)需要检测不同实验设计中的染色质接触。
一般来说,多重GAM的性能与原始GAM类似,但可能需要更多数量的核图谱来检测最弱的接触(包括染色体间接触),或以最高的基因组分辨率。
2.2识别差分和公共触点在上一个实验中,我们发现了基因组分辨率的差异性,为了能更好的识别基因之间的差分,我们还需要去测试他们之间的公共触点。
与Hi-C相比,我们在GAM更大的基因组距离上检测到了更多的接触,例如超级增强子之间的兆碱基范围接触,这已通过单细胞荧光原位杂交实验证实。
Hi-C和GAM数据的计算机模拟表明,在捕获真实距离方面,GAM比Hi-C表现更好(Spearman相关性:Hi-C为-0.89,GAM为-0.99)。
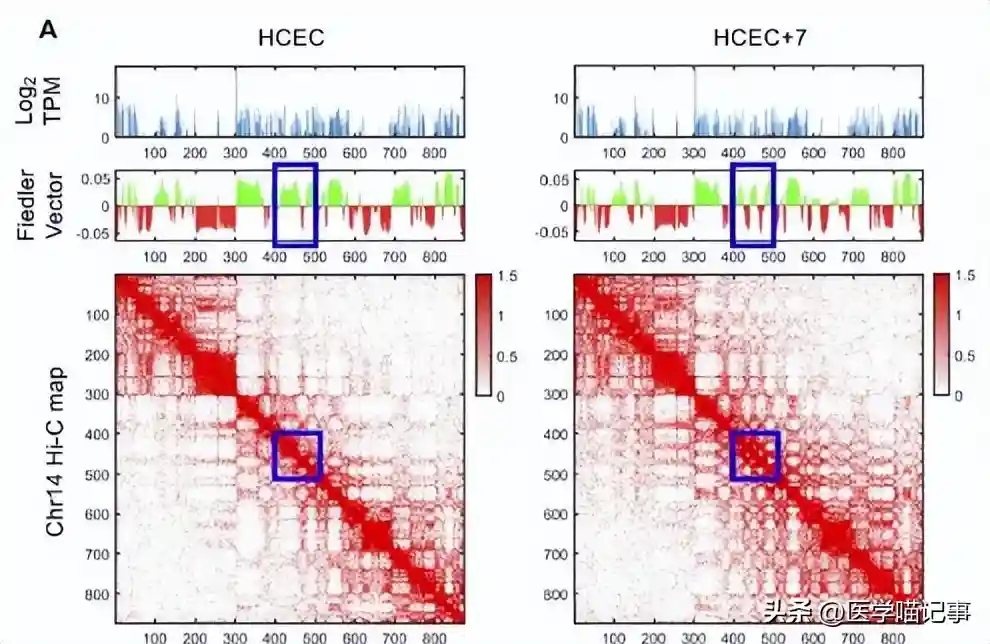
为了更公正地比较GAM和Hi-C之间的全基因组差异,我们开发了一种直接比较两种方法衍生的矩阵的方法。
我们分析考虑了间隔≤4Mb的基因座之间的接触,因为Hi-C的保真度随着基因组距离的增加而降低。
至关重要的是,选定的基因组长度尺度在染色质接触图谱的大多数应用中都很有用,足以检测增强子-启动子接触。
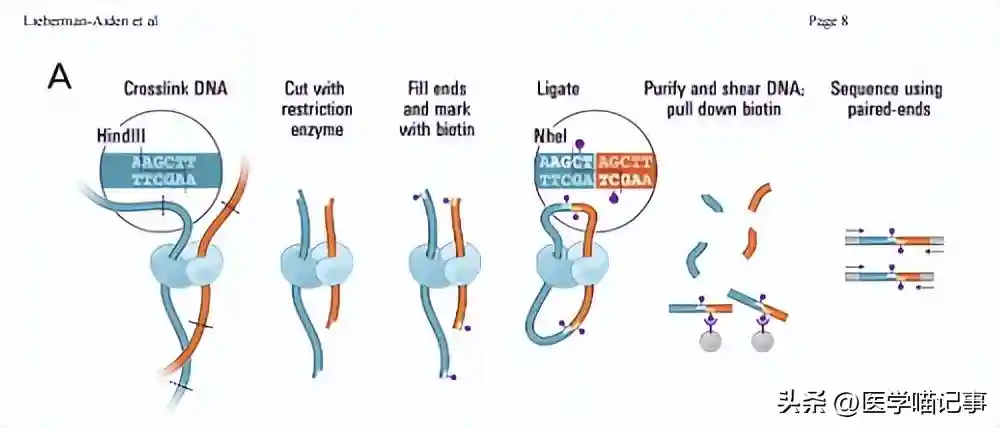
随着实验的继续,我们发现,由于GAM和Hi-C数据具有不同的数值分布,我们首先对两个数据集应用基于距离的z分数变换以解决距离衰减问题。
然后,我们从归一化矩阵中减去这两个矩阵,并提取最显著的接触,即两个矩阵之间的差异大于拟合正态分布定义的5%极值的接触。
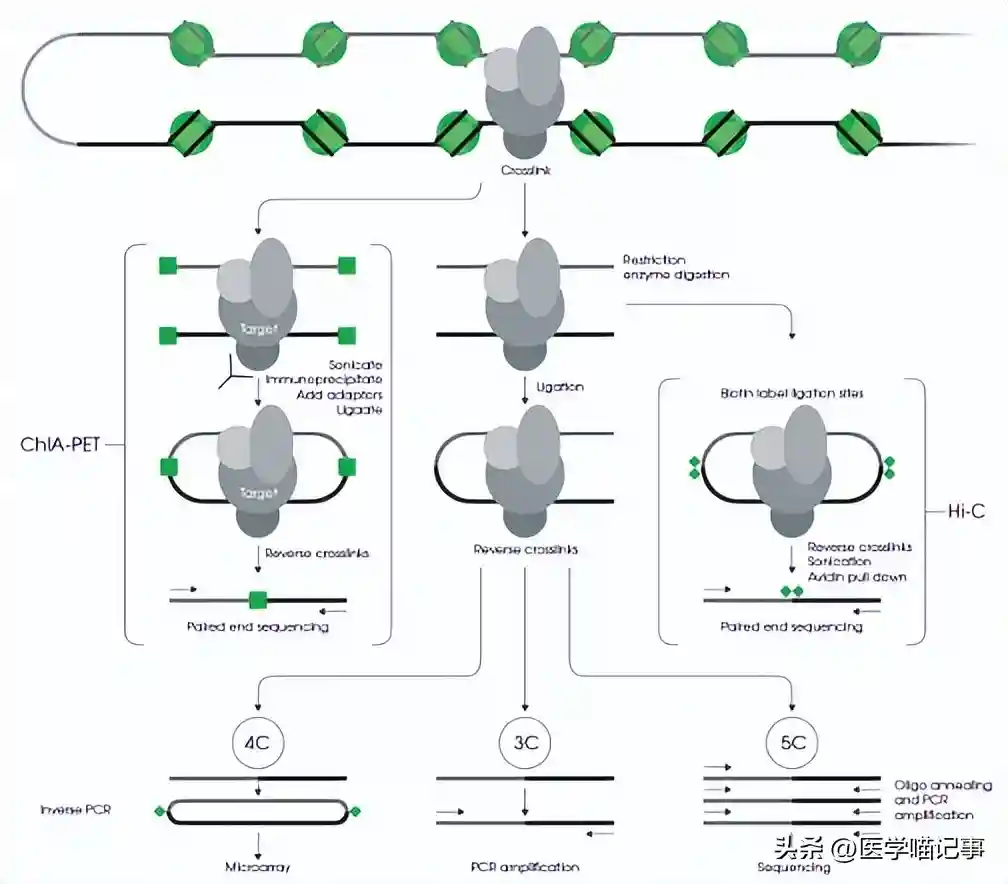
我们还验证了关于GAM特定接触和Hi-C特定接触具有相似的距离衰减,并且大多数也通过替代标准化方法发现。
不仅如此,在所选的GAM特定接触在GAM中同样具有高强度,在Hi-C中具有低强度,对于Hi-C特定接触而言反之亦然。
此外,我们确定了两种方法是否差异地检测到使用GAM数据的SLICE或使用Hi-C数据的Fit-Hi-C捕获的最显著接触。
有意思的是,我们发现虽然最强的GAM接触检测到一定比例的Fit-Hi-C接触,但最强的Hi-C接触却被GAM数据中检测到的最显著的SLICE接触严重耗尽。
最后,我们研究了Hi-C或GAM特异性接触所涉及的基因组窗口的可检测性,发现GAM特异性接触往往源自可检测性最强的窗口,而Hi-C特异性接触往往涉及较少的连接事件。
三、实验结果在分析了GAM和Hi-C特异性接触中特定的基因组特征的显著富集后,我们进一步研究了这两种方法是否存在难以检测到某些特征。
为了更好的发现潜在的盲点,我们又开发了一种计算方法,用于评估每个基因组窗口形成的GAM特异性、Hi-C特异性和强公共接触的数量,并探究特定基因组区域是否更倾向于参与GAM特异性或Hi-C特异性接触,或者是否相反。
令人惊讶的是,我们发现这些盲点窗口相当普遍,如方法特异性接触的耀斑所示。
此外,盲点窗口通常在线性基因组的特定区域聚集。
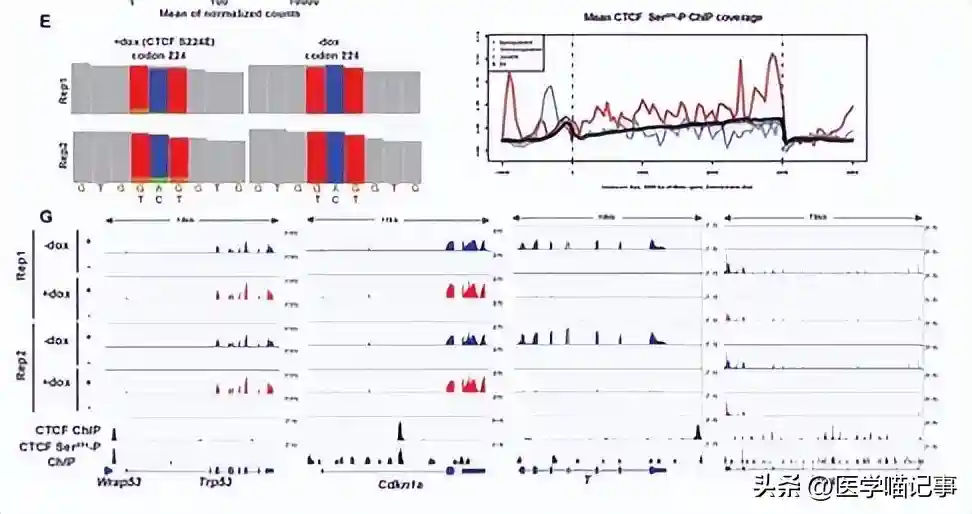
为了深入了解GAM或Hi-C特异性接触中代表性不足的基因组区域的特性,我们选取了方法特异性或强常见接触的前十分位数中的基因组窗口。
我们发现形成许多GAM特异性接触的基因组窗口(我们称之为GAM首选区域)比形成许多Hi-C特异性接触的区域(称为Hi-C首选区域)更为常见,而这些GAM首选区域更频繁地与核纤层相关。
此外,GAM偏好区域通常富集了CTCF、p300、某些小鼠胚胎干细胞转录因子、RNA聚合酶II(特别是延伸型S2p形式)、增强子和超级增强子,并且通常被归类为区室A。
相比之下,Hi-C偏好区域显示出异染色质相关组蛋白标记H4K20me3或H3K9me3的轻微富集,并且更常被分类为区室B。
所有考虑的基因组特征的轨迹也在8号染色体的80Mb区域内显示在基因组浏览器可视化中,它们在同一基因组窗口内的共现突显了GAM首选区域中CTCF、转录活性特征(包括超级增强子)的存在。
四、结语在本次论文中,我们通过使用SLICE计算了位于同一染色体上的三个1Mb窗口的所有可能集合的相互作用概率,以研究基因细胞之间的相互作用复杂性和方法特定盲点之间的关系。
有趣的是,即使在同一区室内进行比较,GAM偏好的区域也比常见或Hi-C偏好的区域形成更多的三联体。
这与我们之前识别长链超级增强子和活跃转录基因组区域之间的染色质接触的研究结果一致。
这些结果表明细胞之间存在丰富的染色质接触,其中许多活性区域同时相互作用,这通常被基于连接的方法所忽视,但很容易被GAM和FISH检测到。
综上所述,在Hi-C数据中,形成许多三联体(即更复杂)的区域具有较低的接触强度。
相反,形成很少三联体的区域在Hi-C中具有更高的接触强度,证明复杂性解释了Hi-C或GAM测量的接触频率的一些差异。
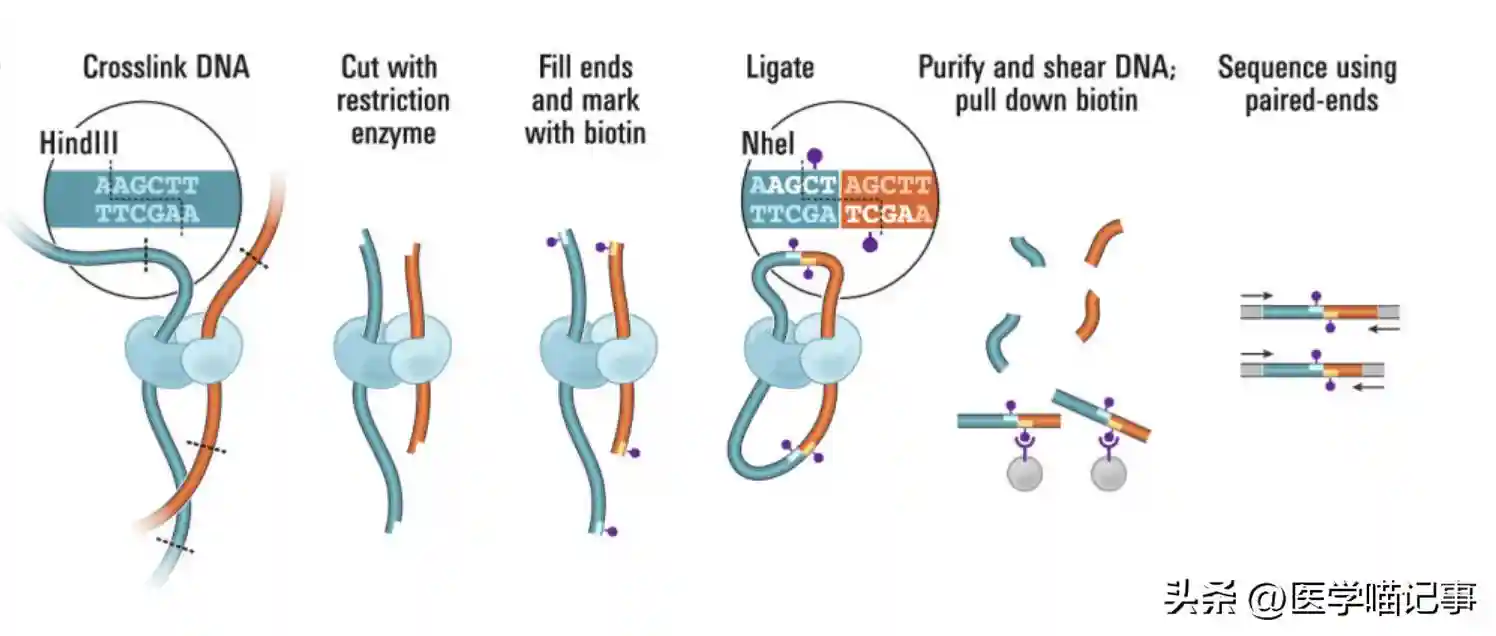

这也说明了我们提供的方式确实在一定程度上降低了全基因组范围内的染色质接触图的时间成本。
不过值得注意的是,这种实验的方式也会一定程度破坏了仅基于成对接触的传递性来预测Hi-C复杂相互作用形成的尝试。

.jpeg "月度汇总 | 2024年9月中国及国际化妆品法规动态")

